The Kuroda Laboratory was established in 2004. The laboratory's field of research lies at the interface between biology, bioinformatics, biophysics. In particular, we aim to elucidate protein-related biological phenomena at the atomic and molecular level and to apply this knowledge to bio-industrial and drug discovery research. Specifically, we are engaged in the design of subunit vaccines, analysis and control of immune responses induced by protein aggregation, and research on mini-antibodies and proteins that inhibit cancer cell proliferation.
Controlling Protein Solubility and Aggregation
We are developing a technology to control the solubility or aggregation of target proteins without changing their function or structure. This technology is based on the simple idea of genetically adding a few hydrophilic amino acid residues to the end of a low-solubility protein. This technology is named SEP (Solubility Enhancement Peptide tag). We are also developing peptide tags to produce aggregates of desired sizes by adding hydrophobic amino acids at the end of the protein (SCP tag for Solubility Controlling Peptide tag). The following features are noteworthy compared to conventional technologies for increasing or decreasing solubility.
- No solvent adjustment is required. SEP and SCP tags can thus be used in vitro and in vivo, which is especially advantageous when producing natively folded proteins in bacteria, especially in E. coli.
- As an example of application, we improved the solubility of TEV (Tobacco Edge Virus) enzyme, which is routinely used for biochemical purification of proteins. Our "super-TEV” enzyme is active under a broader range of conditions than commercial TEV. Furthermore, it is estimated that the price of Super-TEV produced in the lab is less than 1/100 of the commercial product. (A plasmid of our Super-TEV plasmid is free available from ADDGENE)
- By improving the solubility of Gaussia luciferase (GLuc), a luminescent enzyme from sea firefly (Gaussia), natively folded Gluc with bioluminescent activity was expressed in E. coli. Without the SEP tag, Indeed, native GLuc cannot be produced in E-coli because of the missshuffling of its 8 cysteins. The SEP-tagged luciferase is much less costly and faster to produce than the conventional Gluc, produced in insect cells. (distributed from ADDGENE)
- We are currently working on solubilizing an antibody fragment called ScFv.
- In summary, the SEP tag is a versatile technology that succeeded in solubilizing many proteins with molecular weights of a few thousand to up to 50,000.
- Wu Nan, Saotome Tomonori, Unzai Satoru, Kuroda Yutaka* and Yamazaki Toshio *, Solution structure of Gaussia Luciferase with five disulfide bonds and identification of a putative coelenterazine binding cavity by heteronuclear NMR, Scientific Reports volume 10, Article number: 20069 (2020)
- Nautiyal Kalpana and Kuroda Yutaka, A SEP tag enhances the expression, solubility and yield of recombinant TEV protease without altering its activity, N Biotechnol.; 42:77-84 (2018)
- Kato Atsushi, Maki Kosuke, Ebina Teppei, Kuwajima Kunihiro, Soda Kunitsugu and Kuroda Yutaka*, Mutational analysis of protein solubility enhancement using short peptide tags, Biopolymers, 85:12-18 (2007)
- Patent No. 5273438, Yutaka Kuroda, Naoshige Izumikawa, Jun Kato, Shihori Sotani,: "Method for Calculating Solubility of Peptides, and Method for Designing Peptide Tags and Synthesizing Proteins Using the Method", filed January 11, 2008, registered May 24, 2013, patent applicant: National University Corporation Tokyo University of Agriculture and Technology
Biological and Physiological Effects of Protein Aggregation
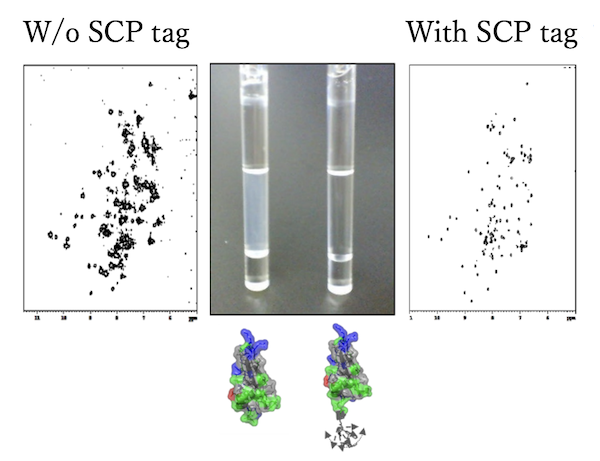
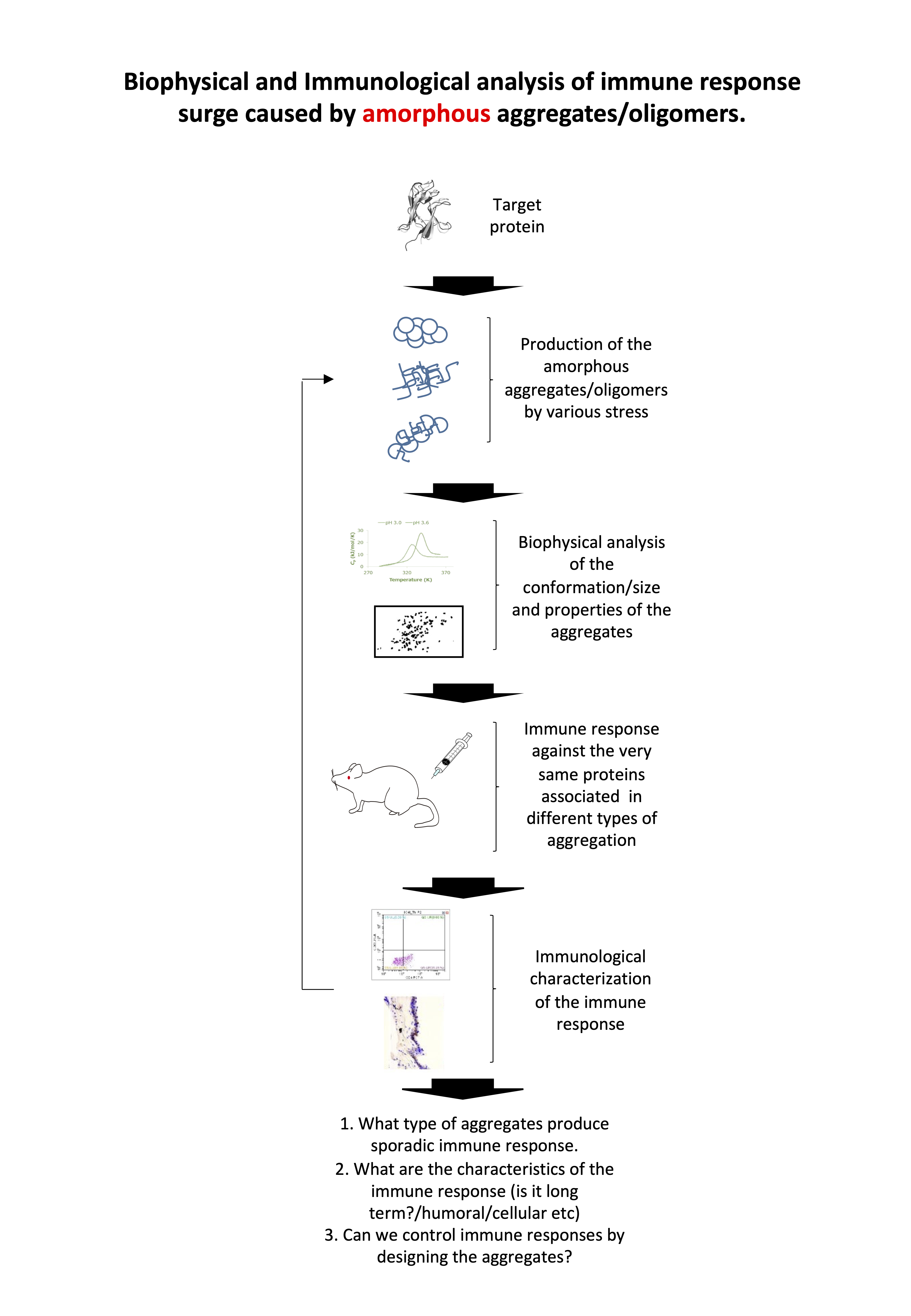
In recent years, the association of protein aggregation to several physiological disorders besides the well-known neurodegenerative diseases has become evident. In particular, we have shown that small proteins, which usually exhibit low immunogenicity, can induce a strong immune response upon aggregation. Indeed, based on several clinical observations, aggregation has long been suspected to be a factor in the sudden induction of an immune response against therapeutic proteins, prompting the release of an FDA guidance on therapeutic proteins (FDA Guidance, 2014). However, few experiments have directly demonstrated the relationship between aggregation and immunogenicity. The reasons include a lack of characterization of the physical properties of amorphous aggregation (aggregation) and the lack of techniques for producing protein aggregates with desired characteristics that are not mature. We are thus developing techniques for enhancing the immunogenicity of proteins by using SCP tags. This technique consists in oligomerizing the proteins into aggregates with specific sizes and physical properties, thereby enhancing the immune response through the cross-linkage of B cell receptors. This research will help elucidate the mechanism of immune response enhancement and enable the control of immunogenicity using SCP tags. We are currently working on applying the SCP tag to increase the immunogenicity of recombinant viral protein domains. Indeed, the low immunogenicity of recombinant proteins is an issue in vaccine development. Using our SCP tags to increase the immunogenicity of recombinant proteins would enormously increase their suitability as a vaccine seed.
- Subbaian Brindha and Kuroda Yutaka, A Multi-Disulfide Receptor-Binding Domain (RBD) of the SARS-CoV-2 Spike Protein Expressed in E. coli Using a SEP-Tag Produces Antisera Interacting with the Mammalian Cell Expressed Spike (S1) Protein. Int. J. Mol. Sci. 23, 1703 (2022)
- Golam Kibria M., Akazawa-Ogawa Y, Hagihara Y, Kuroda Y. *, Immune response with long-term memory triggered by amorphous aggregates of misfolded anti-EGFR VHH-7D12 is directed against the native VHH-7D12 as well as the framework of the analogous VHH-9G8..Eur J Pharm Biopharm. 7: S0939-6411(21) 00128-4(2021)
- Rahman N, Islam MM, Unzai S, Miura S, Kuroda Y., Nanometer-size aggregates generated using short Solubility Controlling Peptide tags do increase the in vivo immunogenicity of a non-immunogenic protein., Mol Pharm. 2020 Mar 31. May 4;17 (5):1629-1637
- Islam MM, Miura Shiho, Hasan MN., Rahman Nafsoon, Kuroda Yutaka*, Anti-Dengue ED3 long-term immune response with T-cell memory generated using Solubility Controlling Peptide tags, Frontiers in Immunology, 2020, Mar 17;11:333
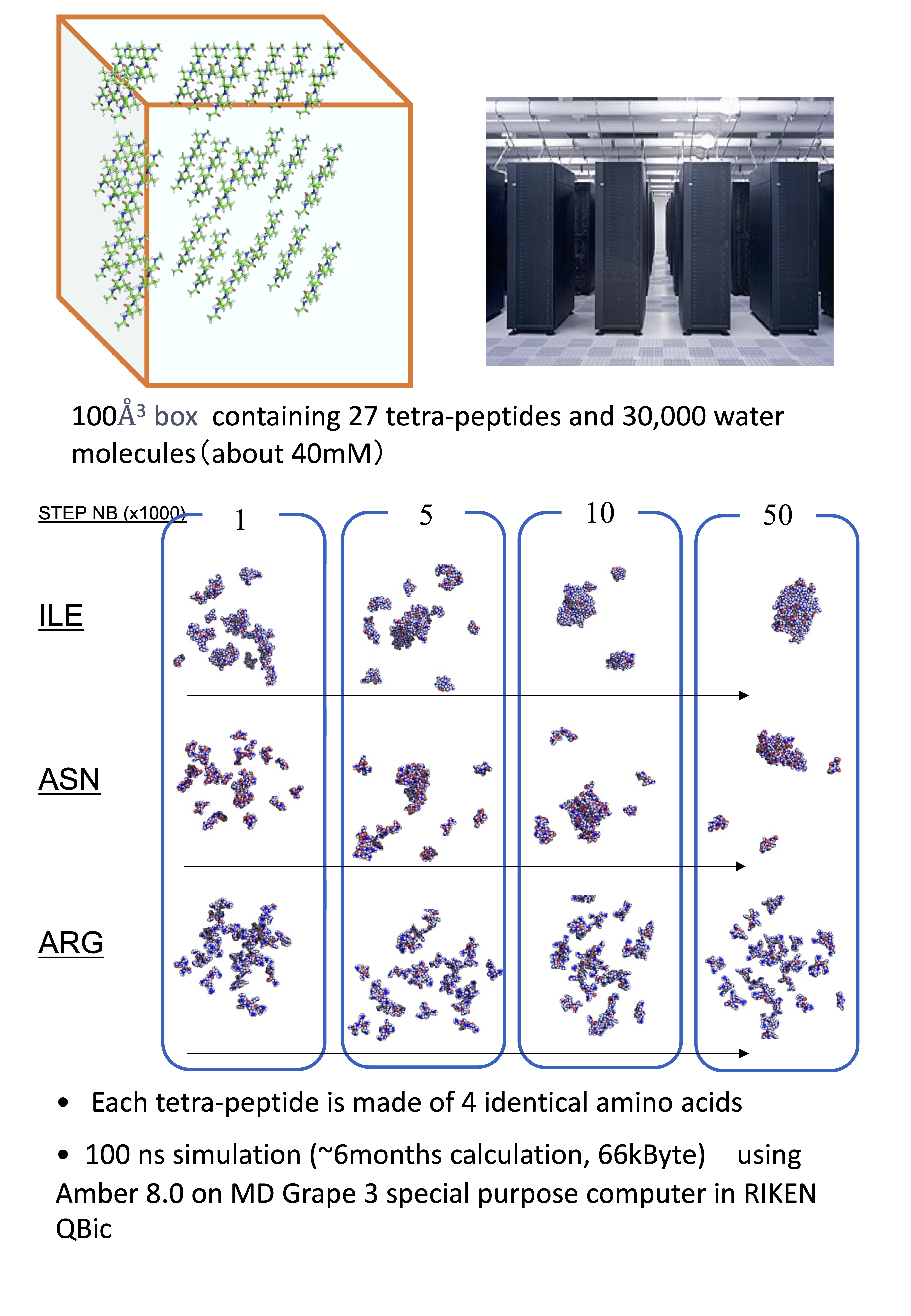
Bioinformatics: Predicting Protein Solubility
The biophysical properties of non-amyloid aggregates (amorphous aggregates) remain poorly characterized. However, recent studies have revealed that amorphous aggregates affect physiological functions. This prompted us to use bioinformatics methods for investigating whether the solubility and aggregation of proteins and peptides can be reproduced using computational methods, including molecular dynamics (MD), Brownian molecular dynamics (BD), and lattice models coupled withMonte Carlo methods. Recently, in collaboration with RIKEN and using the MD-GRAPE supercomputer, we have shown that the relative solubility of amino acids is well estimated by standard MD simulation of a multi-tetrapeptide system. Additionally, we used BD to predict/design the solubility of the above Super-TEV.
- Tambi Richa, Morimoto Gentaro, Kosuda Satoshi, Taiji Makoto, Kuroda Yutaka*, Large-scale all-atom molecular dynamics alanine-scanning of IAPP octapeptides provides insights into the molecular determinants of amyloidogenicity, Scientific Reports, 9(1):2530 (2019)
- Kuroda Yutaka*, Suenaga Atsushi, Sato Yuji, Kosuda Satoshi and Taiji Makoto, All-atom molecular dynamics analysis of multi-peptide systems reproduces peptide solubility in line with experimental observations, Scientific Reports 6, Article number: 19479 (2016 Jan28)
- Nautiyal Kalpana, Kibria Md Golam, Ogawa-Akazawa Yoko, Hagihara Yoshihisa, Kuroda Yutaka*, Design and assessment of an active anti-epidermal growth factor receptor (EGFR) single chain variable fragment (ScFv) with improved solubility, Biochemical and Biophysical Research Communications:508(4):1043-1049 (2019)
- Patent No. 5273438, Yutaka Kuroda, Naoshige Izumikawa, Jun Kato, Shihori Sotani, "Method for Calculating Solubility of Peptides, and Method for Designing Peptide Tags and Synthesizing Proteins Using the Method", filed January 11, 2008, registered May 24, 2013
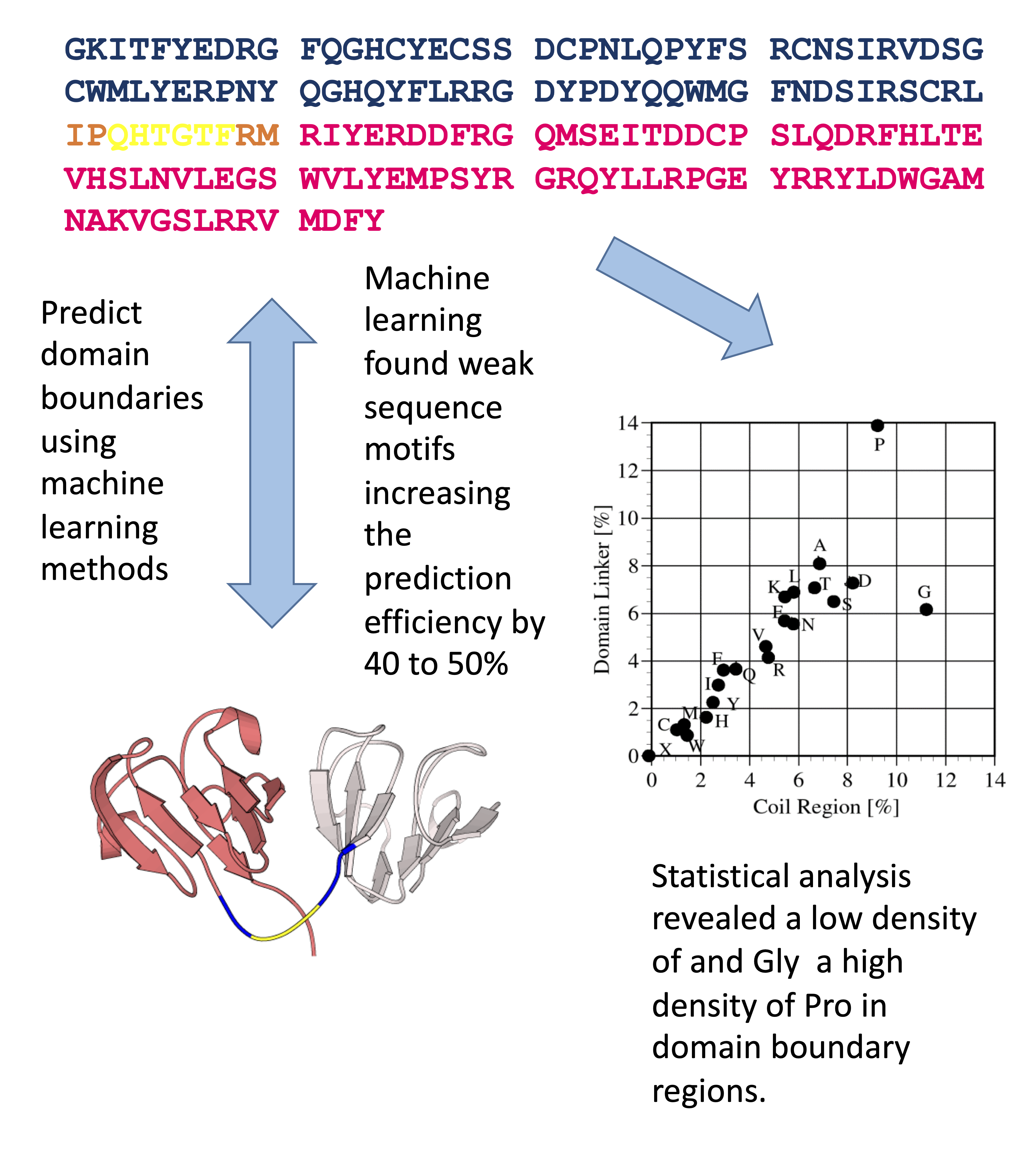
Predicting Protein Domain Boundaries Using machine learning
We have used machine learning (SVM, neural networks) to predict the domain boundaries of large proteins. Domain regions, which can fold and often exhibit biological functions in isolation, are basic units of proteins. Prediction of domain regions from amino acid sequences is helpful for proteome analysis because it provides a method to dissect proteins into domains which can be rapidly analyzed. We have developed a support vector machine (SVM a type of machine learning) that efficiently identifies domain regions of novel proteins from their amino acid sequence information and made it available on the Internet (pls provide link) The prediction rate of this machine was nearly 60%, and it was the best predictor in the domain prediction category of CASP8 (International Contest for the Prediction of Protein Structures, held in 2009).
- Tambi Richa, Ide Shoichi, Ebina Teppei, Kuroda Yutaka, Fast H-DROP: A thirty times accelerated version of H-DROP for interactive SVM-based prediction of helical domain linkers", Journal of Computer-Aided Molecular Design (2):237-244 (2017)
- Ebina Teppei, Toh Hiroyuki, Kuroda Yutaka*, DROP: an SVM domain linker predictor trained with optimal features selected by random forest, Bioinformatics, 27(4):487-94 (2011)
- Patent No. 4213034, Yutaka Kuroda, Satoshi Miyazaki, Takanori Tanaka, Shigeyuki Yokoyama, "Method for predicting domain linkers of proteins", Patent Application 2003-538962, filed September 25, 2002 (2001), Patent applicant: RIKEN
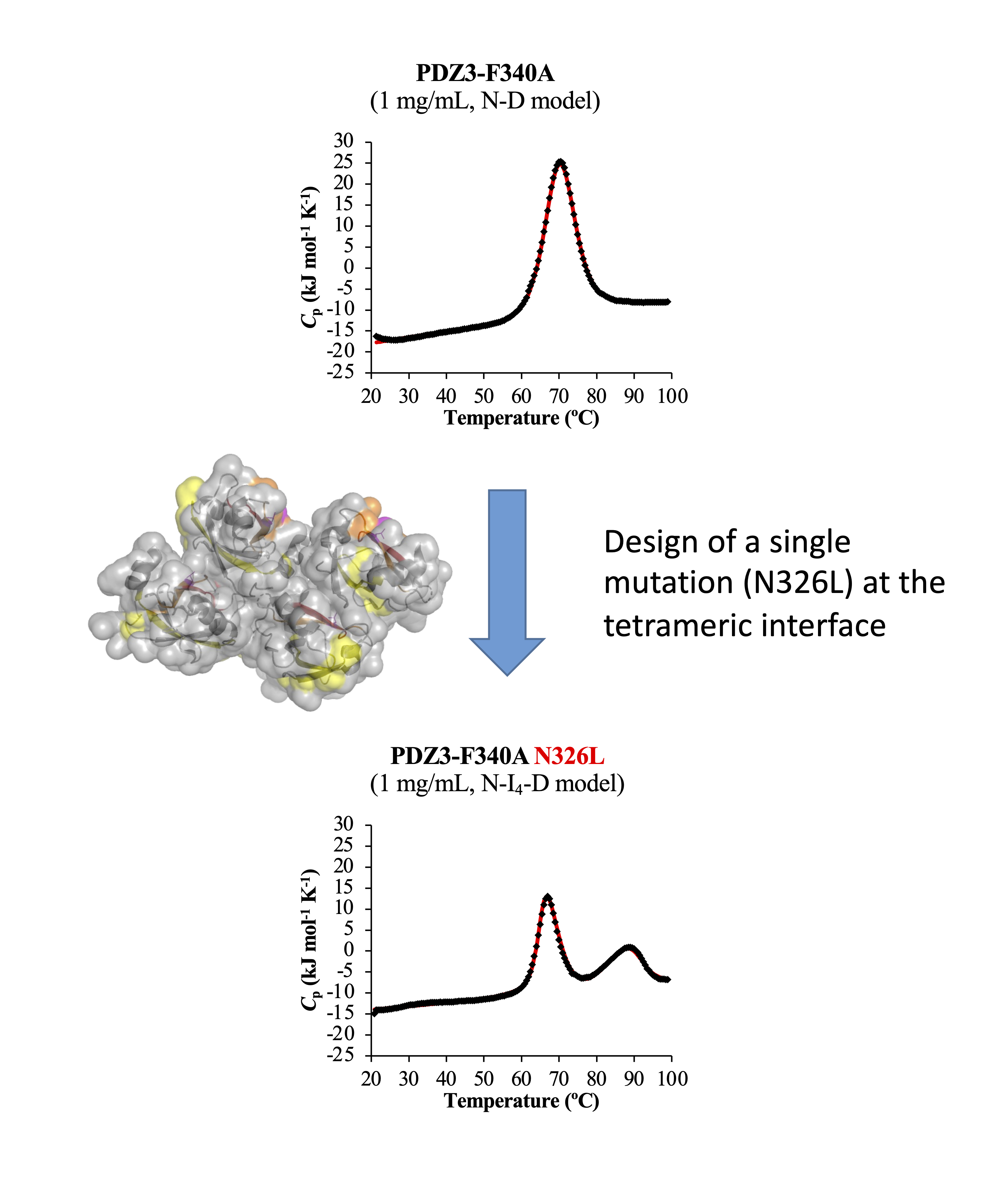
Protein Folding and Stability Research
How proteins fold into their native three-dimensional structure is a fundamental question that has not been answered despite many efforts. Proteins, such as enzymes and antibodies, function by folding their native structures (with some exceptions, such as natural denatured regions). Upon biotechnological and therapeutical usage, it is desirable, if not necessary, for the protein to maintain their native structure, not only at room temperature but sometimes at high temperatures. However, the design principles for stabilizing a protein structure are not fully established and are still a subject of research. For example, in collaboration with Nagaoka Technological University, we recently discovered that hydrophobic amino acids exposed on the surface of globular proteins induce the formation of oligomers at high temperatures. Unexpectedly, when the temperature was increased further, the proteins did not aggregate, but rather the oligomers dissociated into the denatured monomeric state, indicating the existence of a new intermediate (thermodynamic) state during the thermal denaturation of globular proteins. We coined this intermediate state the reversible oligomerization state (RO). Furthermore, the results suggested that this RO is an intermediate for amyloid formation. Amyloid is thought to be the causative molecule of many neurodegenerative diseases, and we thus hope that our basic research on RO may lead to new therapies.
- Onchaiya S., Saotome T., Mizutani K., Martinez JC., Tame JRH., Kidokoro SI., Kuroda Y., Reverse Engineering Analysis of the High-Temperature Reversible Oligomerization and Amyloidogenicity of PSD95-PDZ3. Molecules. 2022 Apr 28;27(9):2813
- Huang YJ., Zhang N., Bersch B., Fidelis K., Inouye M., Ishida Y., Kryshtafovych A., Kobayashi N., Kuroda Y., Liu G., LiWang A., Swapna GVT., Wu N., Yamazaki T., Montelione GT., Assessment of prediction methods for protein structures determined by NMR in CASP14: Impact of AlphaFold2. Proteins. 2021 Dec;89(12):1959-1976
- Saotome T., Onchaiya S., Brindha S., Mezaki T., Unzai S., Noguchi K., Martinez JC., Kidokoro SI., Kuroda Y., Blocking PSD95-PDZ3's amyloidogenesis through point mutations that inhibit high-temperature reversible oligomerization (RO). FEBS J. 2021
- Tomonori Saotome, Maxime Doret, Manjiri Kulkarni, Yin-Shan Yang, Philippe Barthe, Yutaka Kuroda and Christian Roumestand*Folding of the Ig-like domain of the envelope protein from dengue virus analyzed by high hydrostatic pressure NMR at a residue-level resolution Biomolecules. 9,309, 2019 Jul 26;9(8)
- Mohammad, M Islam, Kei Kobayashi, Shun Ichi Kidokoro, Yutaka Kuroda*, Hydrophobic surface residues can stabilize a protein through improved water-protein interactions., FEBS J. 2019 Jun 7
- Islam M M, et al, Crystal structure of an extensively simplified variant of bovine pancreatic trypsin inhibitor in which over one-third of the residues are alanines, Proc. Natl. Acad. Sci. USA, 105:15334-15339 (2008)