大気環境保全技術研修マニュアルより(平成10年3月、海外環境協力センター発行)
大気汚染物質の生成機構
4. 2.1 はじめに
大気汚染物質には、人間活動によって直接放出されている一次汚染質と、一次汚染質が大気中で反応して生成する二次汚染質があることはよく知られているが、本章では二次汚染物質の生成・変質の機構について、特に光化学オゾンの生成、炭化水素の関与する大気化学反応、および酸性雨に関与する反応について解説することとしたい。
4.2.2 オゾン生成の光化学反応
二次汚染物質の生成・変質の過程に関与する反応はおもに一次汚染質としての窒素酸化物(NOx)と炭化水素類(HC)に太陽光のエネルギーが加わって起こるいわゆる光化学スモッグの生成反応である。NO(一酸化窒素)とNO2(二酸化窒素)からなる、NOxは発生源の偏りや、大気中の寿命が短いことなどからその濃度はかなりの幅を持っていて、一般に都市域では20〜500ppb (ppbはppmの1/1000)、田園地域で1〜10ppb、海洋上などのバックグラウンドでは0.05〜0.2ppb程度の濃度である。
NOxは大気中における反応性が非常に高く、従ってその寿命は短い。大気中のほとんどの反応に直接、間接に関与して重要な役割を果たしているが、特に対流圏大気中でのオゾン生成に関しては、都市における光化学スモッグ事件を契機として明らかにされたように、NOxはオゾンの生成に主要な役割を果たしている。対流圏のオゾンの生成源となり得るのはNO2だけであり、この意味からNOxの存在は非常に重要である。
対流圏大気中に微量のNOxが存在していれば、これに光が当たって、式(1)、(2)に示すように、NOの生成を介してオゾンが生成する。
NO2 + hν → O(3P) + NO (1)
O(3P) + O2 → O3 (2)
ここで、O(3P)は基底状態の酸素原子を表す。ところがこれだけだと、式(1)で副成したNOによりオゾンが破壊される反応(3) が起こるので、オゾン濃度は定常状態に達してしまい、極端な高濃度にはならない。
O3 + NO → NO2 + O2 (3)
ところが大気中に炭化水素類が存在すると、NOが炭化水素類の反応によって生成するHO2・やRO2・といった過酸化ラジカルによって、オゾンを破壊することなくNO2に戻されるため(反応4)、
NO + HO2・ or RO2・ → NO2 + ・OH or RO・ (4)
(Rはアルキル基等炭化水素に由来するグループ)
このNO2が再び(1),(2)の反応を経てオゾンを生成する。その結果、光が強い条件の下では、オゾンが高濃度になるのである。
対流圏大気中の化学反応は、・OHラジカルによって開始される場合が多い。特に、オゾンと反応しないアルカン類や芳香族炭化水素類はほとんど・OHラジカルとの反応によって消滅して行く。対流圏大気中の・OHは主に次式の反応で生成するが、NOxや炭化水素を含むラジカル連鎖反応によって(4)のように再生され再び反応にあずかることになる。
O3 + hν → O2 + O(1D) (5)
O(1D) + H2O → 2・OH (6)
ここで、O(1D)は励起状態の酸素原子である。基底状態の酸素原子O(3P)は水とは反応しない。
図1
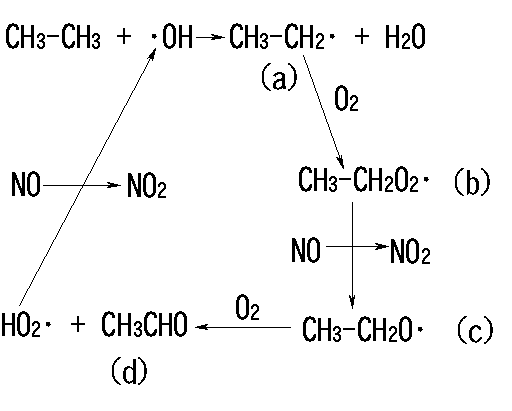
4.2.3 炭化水素の反応
図1に示したのは代表的な炭化水素のアルカンの典型的な反応である。このような連鎖反応によって、炭化水素に含まれる炭素は徐々に二酸化炭素まで酸化されて行き、NO→NO2の変換が起こってオゾンが蓄積されることは上に述べた通りである。アルキル鎖が長くなるかまたは大きくなると、(b)のタイプの生成物(パーオキシラジカル)からは図1に示したNOの酸化だけでなく、式(7)に示すようなNOの付加と転位反応による硝酸エステル(RONO2)の生成も重要になってくる。
RO2・ + NO → [ROONO] → [RONO2]* → RONO2 (7)
また生成物(c)のアルコキシルラジカル(RO・)は図1に示したように主に酸素と反応するが、十分な濃度のNOxが存在すると、式(8)のようにNOxの付加反応により、硝酸エステルや亜硝酸エステルを生成する。
RO・ + NO2 or NO → RONO2 or RONO
(8)
亜硝酸エステルは可視光によって容易に分解され、アルコキシルラジカルを生成して、図1のような連鎖反応を開始することができるので、夜間にオゾン−オレフィン反応やNO3ラジカルと炭化水素の反応等にともなって式(8)により亜硝酸エステルが蓄積されていると、光化学反応の重要な開始剤になることが知られている1)。
NOxを含むもう一つ重要な反応生成物は、いわゆるPANと呼ばれている化合物である。PANとは、もともとはperoxyacyl nitrate(RC(=O)OONO2)の総称であったが、現在ではその中で最も重要なperoxyacetyl
nitrate(CH3C(=O)OONO2)を指すことが多い。その最も重要な生成過程は、図1に示したように、エタン以上の炭化水素の光化学反応で生じた(または一次汚染質として放出された)アセトアルデヒドと・OHの反応によって開始される、以下のような過程である。
CH3CHO + ・OH → CH3CO + H2O (9)
CH3CO + O2 → CH3C(=O)OO (10)
CH3C(=O)OO + NO2 → CH3C(=O)OONO2 (11)
PANは光化学オキシダントの一つで、植物に可視傷害を与えたり、人体への被害も知られている毒性の強い物質であるが、大気化学的な見地から、最近では窒素のリザバーとしての役割の重要性が指摘されている2)。NOxの大気中での寿命は非常に短いので、NOxはあまり長距離を移動すること(長距離輸送)はなく、発生源の近傍で反応によって消滅してしまう。しかし、一旦PANとなって、上空の気温の低い所に移るとその寿命は非常に長くなる。例えば、気温20℃でのPANの寿命は1.7時間しかないが(消滅過程は主に(11)の逆反応の熱分解である)、0℃では50時間、-20℃では105日にもなる3)。従って、窒素がPANの形で長距離輸送され、その後分解してNO2を放出し光化学反応に関与することも十分に考えられる。実際、北半球中緯度地帯の対流圏で見られる春先のオゾンの高濃度現象は、冬の間に蓄積・輸送されたPANが気温の上昇とともに分解し、生じたNO2がオゾンの生成源となっているためではないかとの指摘4)もなされている。
4.2.4 対流圏大気の酸性化(いわゆる酸性雨)に関与する大気化学反応
酸性雨現象も地球規模の環境問題の一つとして大きな関心を集めている。雨そのものの酸性化もさることながら、対流圏大気の酸性化が問題であり、雨の酸性化はその結果に過ぎないと考えるべきである。言うまでもなく、対流圏大気の酸性化は、人間が様々な活動によって放出している硫黄酸化物や窒素酸化物が増大していることに起因している。これらの化合物が大気中で酸化されると硫酸や硝酸になり、これが雨に溶け込んで酸性雨となる。
NOxの増加は、上記のように対流圏大気中のオゾンや種々の酸化性ラジカル(・OH、HO2・、RO2・等)の濃度を増加させ、大気の酸化能をも増大させている。これも酸性物質前駆体の増加とあいまって大気中の酸性物質の増加をもたらしている。
酸性物質の生成にはSO2の酸化による硫酸の生成とNOxの酸化による硝酸の生成がある。
SO2の酸化には・OHラジカルによる以下のような気相の均一反応と
SO2 + OH → HOSO2 (12)
HOSO2 + O2 → SO3 + HO2 (13)
SO3 + H2O → H2SO4 (14)
SO2が雲や霧、雨滴などの水に溶けた後の溶液中での反応がいずれも重要である。溶液中の反応では過酸化水素が主要な役割を果たしている5)。
一方、NOxの酸化による硝酸の生成過程は割合単純で、SO2の場合のような液相での酸化反応は、NOxが水に溶けにくいため、ほとんど起こらない6)。主に次の三つの反応によって進行するものと考えられている。
(1)・OHとの反応
硝酸の生成に対しても式(15)に示すように・OHの役割は重要である。
NO2 + ・OH → HNO3 (15)
(2)NO3ラジカルの反応
NO3ラジカルはオゾンとNO2の反応で生成する反応性の高いラジカルである。特に、光のあたらない夜間に炭化水素を含む種々の有機化合物の反応において重要な役割を果たしている。NO3ラジカルは炭化水素やアルデヒド類等の有機化合物と反応することにより(16)式のように硝酸に転換される。
NO3 + RH → HNO3 + R・ (16)
(3)N2O5の反応
N2O5は、 HNO3とNO2の反応で生成し、含窒素汚染質の一つとして夜間に起こる反応に重要な役割を果たしている。N2O5はいわば硝酸の無水物であり、(17)式のような加水分解が可能である。ただし、気相均一系における水蒸気との直接反応の速度定数は2x10-21 cm3molecule-1s-1以下7)であり、重要ではない。しかし、この反応によるNOxの消滅過程または硝酸の生成過程は表面が存在するとで加速されるものであり、野外における硝酸の生成過程として重要である。
NO3 + NO2 → N2O5 (17)
文献
1)B.J. Finlayson-Pitts and J.N. Pitts, Jr., "Atmospheric Chemistry",
John Wiley & Sons, Inc., New York, (1986), p.429.
2)H.B. Singh and P.L. Hanst, Geophys. Res. Lett., 8, 941 (1981).
3)B.J. Finlayson-Pitts and J.N. Pitts, Jr., "Atmospheric Chemistry",
John Wiley & Sons, Inc., New York, (1986), p.550.
4)S.A. Penkett and K.A. Brice, Nature (London), 319, 655 (1986).
5)J.G. Calvert, ed., "SO2, NO and NO2 Oxidation Mechanisms: Atmospheric
Considerations", Butterworth, Boston, 1984.
6)S.N. Pandis and J.H. Seinfeld, J. Geophys. Res., 94, 1105 (1989).
7)W.B. De More, S.P. Sander, D.M. Golden, M.J. Molina, R.F. Hampson, M.J.
Kurylo, C.J. Howard, and A.R. Ravishankara, "Chemical Kinetics and
Photochemical Data for Use in Stratospheric Modeling, Evaluation Number
9", JPL Publication 90-1, pp. 217 (1990).